
Somil Gupta
Leonard S. Ornstein Laboratory, room 1.73
Princetonplein 1, 3584 CC Utrecht
P.O. Box 80 000, 3508 TA Utrecht
The Netherlands
phone: +31 (0)30 253 3125
secretariat: +31 (0)30 253 2952
e-mail: s.s.gupta@uu.nl
Research
Supervisor: Dr. ir. Marijn van Huis
Promotor: Prof. dr. ir. Marjolein Dijkstra
Funding: FOM
Employed: 15 July 2014 – 14 July 2018
Role of magnetic defects at the water-CdS interface
Over the past decade, CdS nano-heterostructures with other semiconductors and metal co-catalysts have been developed for photocatalytic hydrogen production.[1]
Fig. 1 shows a schematic of the currently proposed mechanism. An understanding of the water-CdS interface is vital for elucidating the near-surface dynamics of water, and for discerning active sites for surface catalysis. To identify the key surface motifs at this interface, we study the molecular interaction of water on the clean and defected surface facets of wurtzite CdS.
The cationic vacancy defects in II-VI bulk semiconductors are known to have a net local spin, resulting from localized holes at the neighboring anions. Using spin-polarised density functional theory we study these vacancies at the most stable CdS surfaces, and study their interaction with a water molecule. We find that the magnetic moment at the cationic vacancy survives at the surface, only when neighboring S-atom relaxations are restricted by the surface geometry. At one of the more stable magnetic defect centres, we observe very strong adsorption of a water molecule. This is in agreement with experimental literature where a strong adsorption of water vapour on CdS surfaces was reported. Such a magnetic surface motif was so far not considered in simulating the water-semiconductor interfaces; our simulations indicate their relevance, and allow a comparison with electronic paramagnetic resonance experiments.
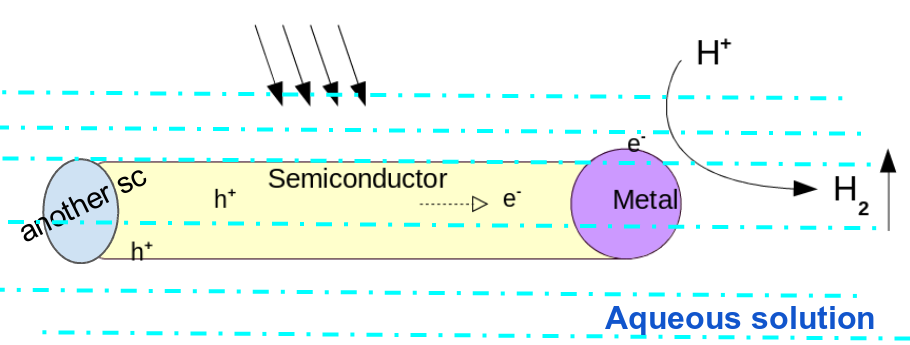
Figure 1: Schematic of the photo-catalytic splitting of water using semiconductor nano-heterostructures
[1] Chen et al., Chemical Reviews 110, 6503–6570 (2010)