
Dr. Laura Filion
Leonard S. Ornstein Laboratory, room 0.22
Princetonplein 1, 3584 CC Utrecht
P.O. Box 80 000, 3508 TA Utrecht
The Netherlands
phone: +31 (0)30 253 2361
secretariat: +31 (0)30 253 2952
e-mail: l.c.filion@uu.nl
Curriculum Vitae: link
Research
Overview
When zoomed in on the nano or micronscale, the natural world is almost unbelievably beautiful: an amazingly rich collection of natural building blocks move collectively to give both structure and function to our world. This self-organization is responsible for diverse phenomena – from the amazing structural colors exhibited in gem opals or in the wings of Morpho butterflies, to the complex dance carried out in cells that allows life to flourish. Mimicking these self-assembly processes to create new materials, designed to exhibit desired and targeted material properties is arguably the holy grail of materials science and a key ingredient in creating the materials that will help us in realizing a sustainable future.
Computer simulations play a key role in this quest: allowing us to study self-assembly processes and giving us insights into how the interactions between the building blocks of a material – whether atoms, molecules, proteins, or colloidal particles – give rise to the final structure.
My research group uses, and develops, a combination of state-of-the-art computational and machine learning algorithms to study self-assembly in soft matter systems – both in and out of equilibrium. We have made significant contributions to our understanding of how defects manifest in colloidal crystals, crystal nucleation, the relationship between structure and dynamics in supercooled liquids, and self-assembly behaviour in active colloids.
Research Highlights:
Improving the prediction of glassy dynamics by pinpointing the local c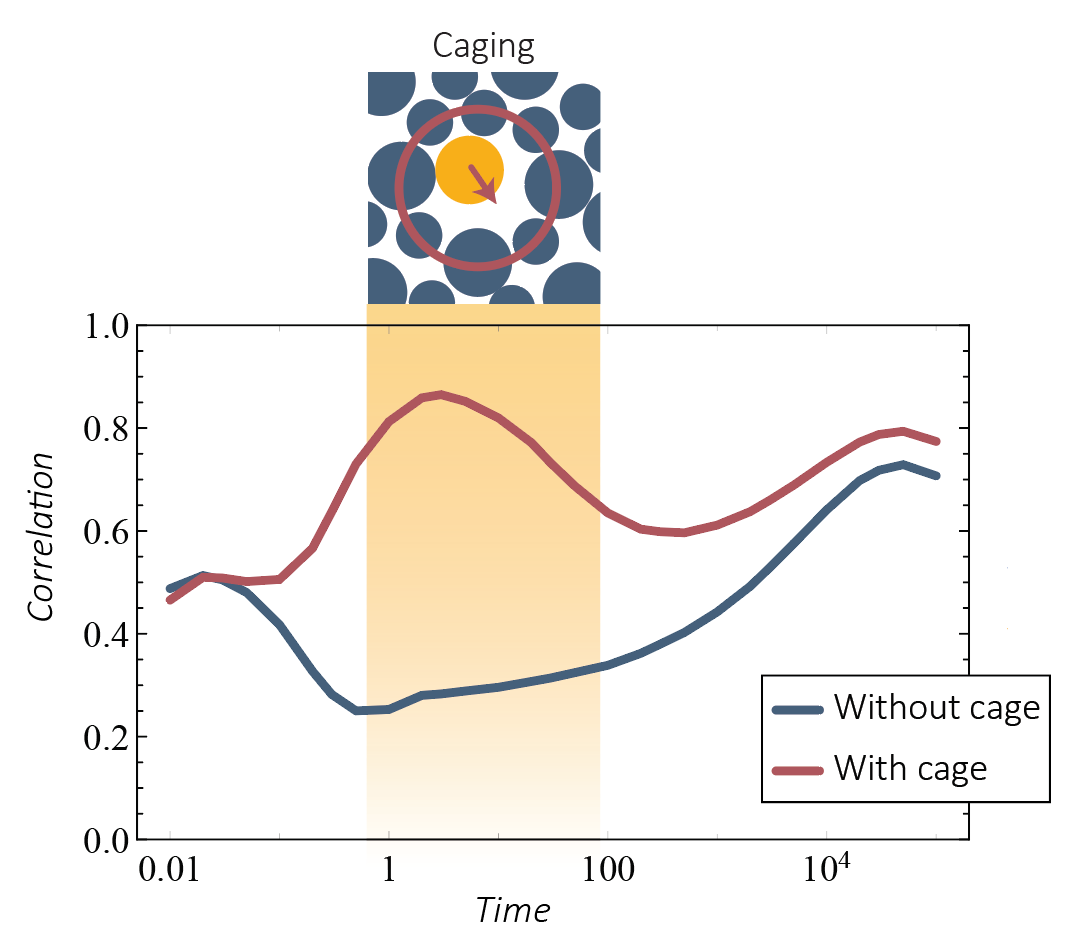 age
age
The relationship between structure and dynamics in glassy fluids remains an intriguing open question. Recent work has shown impressive advances in our ability to predict local dynamics using structural features, most notably due to the use of advanced machine learning techniques. Here, we introduce a method to pinpoint the cage state of the initial configuration—i.e., the configuration consisting of the average particle positions when particle rearrangement is forbidden. We find that, in comparison to both the initial state and the inherent state, the structure of the cage state is highly predictive of the long-time dynamics of the system. Moreover, by combining the cage state information with the initial state, we are able to predict dynamic propensities with unprecedentedly high accuracy over a broad regime of time scales, including the caging regime.
For more information see:
Improving the prediction of glassy dynamics by pinpointing the local cage
Rinske Alkemade, Frank Smallenburg and Laura Filion,
J. Chem. Phys. 158, 134512 (2023).
In search of a precursor for crystal nucleation of hard and charged colloids
The interplay between crystal nucleation and the structure of the metastable fluid has been a topic of significant debate over recent years. In particular, it has been suggested that even in simple model systems such as hard or charged colloids, crystal nucleation might be foreshadowed by significant fluctuations in local structure around the location where the nucleus first arises. We investigate this using computer simulations of spontaneous nucleation events in both hard and charged colloidal systems. To detect local structural variations, we use both standard and unsupervised machine learning methods capable of finding hidden structures in the metastable fluid phase. We track  numerous nucleation events for the face-centered cubic and body-centered cubic crystals on a local level and demonstrate that all signs of crystallinity emerge simultaneously from the very start of the nucleation process. We thus conclude that we observe no precursor for the crystal nucleation of hard and charged colloids.
numerous nucleation events for the face-centered cubic and body-centered cubic crystals on a local level and demonstrate that all signs of crystallinity emerge simultaneously from the very start of the nucleation process. We thus conclude that we observe no precursor for the crystal nucleation of hard and charged colloids.
For more information see:
In search of a precursor for crystal nucleation of hard and charged colloids
Marjolein de Jager, Frank Smallenburg, Laura Filion
J. Chem. Phys. 159, 134902 (2023).
Averaging local structure to predict the dynamic propensity in supercooled liquids
Predicting the local dynamics of supercooled liquids based purely on local structure is a key challenge in our quest for understanding glassy materials. Recent years have seen an explosion of methods for making such a prediction, often via the application of increasingly complex machine learning techniques. The best predictions so far have involved so-called Graph Neural Networks (GNNs) whose accuracy comes at a cost of models that involve on the order of 105 fit parameters. Here, we propose that the key structural ingredient to the GNN method is its ability to consider not only the local structure around a central particle, but also averaged structural features centered around nearby particles. We demonstrate that this insight can be exploited to design a significantly more efficient model that provides essentially the same predictive power at a fraction of the computational complexity, and demonstrate its success by fitting the dynamic propensity of Kob-Andersen and binary hard-sphere mixtures.
For more information see:
Averaging local structure to predict the dynamic propensity in supercooled liquids
Emanuele Boattini, Frank Smallenburg & Laura Filion,
Physical Review Letters 127, 088007 (2021).
Autonomously revealing hidden local structures in supercooled liquids
Few questions in condensed matter science have proven as difficult to unravel as the interplay between structure and dynamics in supercooled liquids. To explore this link, much research has been devoted to pinpointing local structures and order parameters that correlate strongly with dynamics. Here we use an unsupervised machine learning algorithm to identify structural heterogeneities in three archetypical glass formers—without using any dynamical information. In each system, the unsupervised machine learning approach autonomously designs a purely structural order parameter within a single snapshot. Comparing the structural order parameter with the dynamics, we find strong correlations with the dynamical heterogeneities. Moreover, the structural characteristics linked to slow particles disappear further away from the glass transition. Our results demonstrate the power of machine learning techniques to detect structural patterns even in disordered systems, and provide a new way forward for unraveling the structural origins of the slow dynamics of glassy materials.
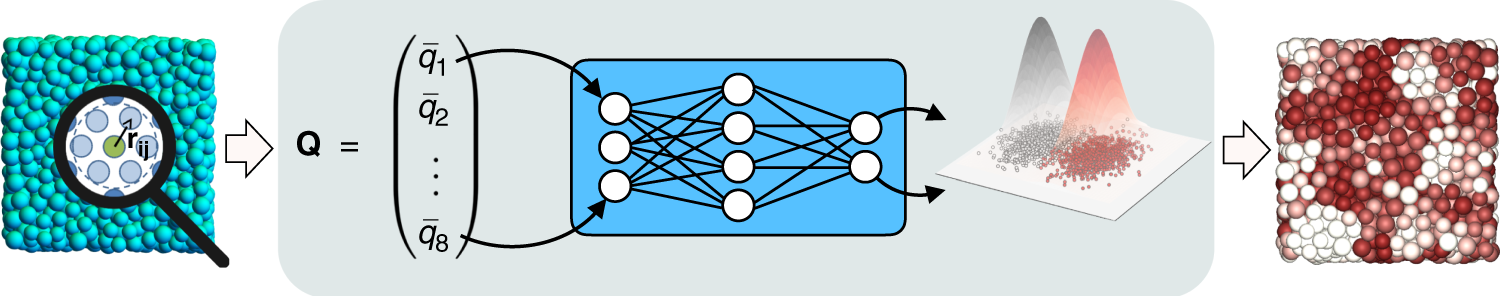
For more information see:
Autonomously revealing hidden local structures in supercooled liquids
Emanuele Boattini, Susana Marín-Aguilar, Saheli Mitra, Giuseppe Foffi, Frank Smallenburg & Laura Filion, Nature Communications 11, 5479 (2020)
Unsupervised learning for local structure detection in colloidal systems
We introduce a simple, fast, and easy to implement unsupervised learning algorithm for detecting different local environments on a single-particle level in colloidal systems. In this algorithm, we use a vector of standard bond-orientational order parameters to describe the local environment of each particle. We then use a neural-network-based autoencoder combined with Gaussian mixture models in order to autonomously group together similar environments. We test the performance of the method on snapshots of a wide variety of colloidal systems obtained via computer simulations, ranging from simple isotropically interacting systems to binary mixtures, and even anisotropic hard cubes. Additionally, we look at a variety of common self-assembled situations such as fluid-crystal and crystal-crystal coexistences, grain boundaries, and nucleation. In all cases, we are able to identify the relevant local environments to a similar precision as “standard,” manually tuned, and system-specific, order parameters. In addition to classifying such environments, we also use the trained autoencoder in order to determine the most relevant bond orientational order parameters in the systems analyzed.
For more information see:
Unsupervised learning for local structure detection in colloidal systems
Emanuele Boattini, Marjolein Dijkstra & Laura Filion,
Journal of Chemical Physics 151, 154901 (2019)
Removing grain boundaries from three-dimensional colloidal crystals using active dopants
 Self-propelled particles, also known as active particles, incessantly convert energy into self-propulsion, and as such are intrinsically out-of-equilibrium. While traditionally such particles occurred solely within the purview of natural systems (e.g. bacteria), recent experimental breakthroughs have led to many novel types of synthetic colloidal swimmers. These systems exhibit a wealth of new phase behaviour, including motility-induced phase separation into dense and dilute phases, giant density fluctuations, and swarming.
Self-propelled particles, also known as active particles, incessantly convert energy into self-propulsion, and as such are intrinsically out-of-equilibrium. While traditionally such particles occurred solely within the purview of natural systems (e.g. bacteria), recent experimental breakthroughs have led to many novel types of synthetic colloidal swimmers. These systems exhibit a wealth of new phase behaviour, including motility-induced phase separation into dense and dilute phases, giant density fluctuations, and swarming.
Moreover, experimental and simulation studies have shown that the dynamics of a passive
system can be altered dramatically by incorporating as little as 1% of active particles into the
system. At these concentrations, the self-propelled particles can be viewed as active “dopants”, which like passive dopants can strongly alter the properties (e.g. dynamics) of the underlying passive system.
We use simulations to explore the behaviour of two- and three- dimensional colloidal (poly)crystals doped with active particles. We show that these active dopants can provide an elegant new route to removing grain boundaries in polycrystals.
For more information see:
Removing grain boundaries from three-dimensional colloidal crystals using active dopants
B. van der Meer, M. Dijkstra and L. Filion, Soft Matter 12, 5630-5635, (2016)
Self assembly of active attractive spheres
One of the most exciting recent developments in the field of colloidal self-assembly is the realization of synthetic, self-propelled colloidal particles. Like living organisms, these “active” colloidal particles convert energy from their environment into directed motion. As a result, these systems are inherently out-of-equilibrium, and are not bound by the laws of equilibrium statistical physics – making them both very intriguing and challenging to study.
 Here we used Brownian Dynamics simulations to examine one of the simplest examples of such an active system: namely a system of active, attractive spheres in three dimensions, where the attraction was modeled using the Lennard-Jones potential. We extracted how the state diagram was affected by the speed with which the particles rotated. Specifically, for slow rotational diffusion, we showed that the gas-liquid coexistence is transformed into a novel percolating network state. Interestingly, this state exhibits high degrees of local orientational alignment of the self-propulsion axis, despite the absence of an aligning force in the model. We also propose a simple mechanism which explains how this alignment and simultaneously rationalizes how activity changes the state diagram.
Here we used Brownian Dynamics simulations to examine one of the simplest examples of such an active system: namely a system of active, attractive spheres in three dimensions, where the attraction was modeled using the Lennard-Jones potential. We extracted how the state diagram was affected by the speed with which the particles rotated. Specifically, for slow rotational diffusion, we showed that the gas-liquid coexistence is transformed into a novel percolating network state. Interestingly, this state exhibits high degrees of local orientational alignment of the self-propulsion axis, despite the absence of an aligning force in the model. We also propose a simple mechanism which explains how this alignment and simultaneously rationalizes how activity changes the state diagram.
For more information see:
Self-assembly of active attractive spheres
V. Prymidis, H. Sielcken, L. Filion, Soft Matter 11, 4158 (2015)
Erasing no-man’s land by thermodynamically stabilizing the liquid-liquid transition in tetrahedral particles
 One of the most fascinating yet controversial hypotheses for explaining the origin of the numerous thermodynamic anomalies characterizing liquid water postulates the presence of a metastable, second-order, liquid–liquid critical point (LLCP) . Located in the so-called ´´no-man´s land´´, where spontaneous crystallization obscures the liquid–liquid phase transition, it is impossible to directly access the LLCP experimentally in order to conclusively prove its existence. Here, we use a simple, single-component patchy-particle model to identify two key ingredients controlling the existence of a LLCP, namely the softness of the interparticle interaction and the flexibility of the bond orientation. We systematically explore the phase behavior of this model, mapping out the competition between crystallization and liquid–liquid phase separation. We show that for certain choices of the interaction parameters, the liquid–liquid phase transition can be made thermodynamically stable, enabling the study of this phenomenon without interference of crystallization at any temperature. Realizing these conditions in soft-matter systems would open up the possibility to experimentally probe liquid–liquid phase transitions, shedding new light on the phase behavior of water and similar tetrahedral liquids.
One of the most fascinating yet controversial hypotheses for explaining the origin of the numerous thermodynamic anomalies characterizing liquid water postulates the presence of a metastable, second-order, liquid–liquid critical point (LLCP) . Located in the so-called ´´no-man´s land´´, where spontaneous crystallization obscures the liquid–liquid phase transition, it is impossible to directly access the LLCP experimentally in order to conclusively prove its existence. Here, we use a simple, single-component patchy-particle model to identify two key ingredients controlling the existence of a LLCP, namely the softness of the interparticle interaction and the flexibility of the bond orientation. We systematically explore the phase behavior of this model, mapping out the competition between crystallization and liquid–liquid phase separation. We show that for certain choices of the interaction parameters, the liquid–liquid phase transition can be made thermodynamically stable, enabling the study of this phenomenon without interference of crystallization at any temperature. Realizing these conditions in soft-matter systems would open up the possibility to experimentally probe liquid–liquid phase transitions, shedding new light on the phase behavior of water and similar tetrahedral liquids.
For more information see:
Erasing no-man’s land by thermodynamically stabilizing the liquid-liquid transition in tetrahedral particles
F. Smallenburg, L. Filion, F. Sciortino, Nature Physics 10, 653 (2014).
Vacancy-stabilized crystalline order in hard cubes
 We examined the effect of vacancies on the phase behavior and structure of hard cubes using event-driven molecular dynamics and Monte Carlo simulations. We found a first-order phase transition between a fluid and a simple cubic crystal phase which is stabilized by an amazingly large number of vacancies: near bulk coexistence the net vacancy concentration is approximately 6.4%, which is more than two orders of magnitude higher than that of hard spheres. Remarkably, we also found that vacancies increased the positional order in the system. A closer examination of the vacancies showed that they were delocalized and extended over several lattice sites. A snapshot of a typical configuration is shown below where the vacancies and the particles which surround the vacancy have been highlighted.
We examined the effect of vacancies on the phase behavior and structure of hard cubes using event-driven molecular dynamics and Monte Carlo simulations. We found a first-order phase transition between a fluid and a simple cubic crystal phase which is stabilized by an amazingly large number of vacancies: near bulk coexistence the net vacancy concentration is approximately 6.4%, which is more than two orders of magnitude higher than that of hard spheres. Remarkably, we also found that vacancies increased the positional order in the system. A closer examination of the vacancies showed that they were delocalized and extended over several lattice sites. A snapshot of a typical configuration is shown below where the vacancies and the particles which surround the vacancy have been highlighted.
For more information see :
Vacancy-stabilized crystalline order in hard cubes,
F. Smallenburg, L. Filion, M. Marechal and M. Dijkstra, PNAS, 109, 17886 (2012),
and a commentary on this article by Daan Frenkel “Colloidal crystals full of invisible vacancies”.